
How can your SaMD pass the clinical evaluation phase with flying colours in EU markets?
Before we dive into clinical evaluation, what is SaMD (Software as a Medical Device) exactly?
A SaMD (Software as a Medical Device) refers to software that is used for a medical purpose, without being part of a physical medical device. For example, it may refer to an application for a computer, mobile phone, or tablet that is intended to treat, diagnose, relieve, monitor, or cure a medical condition or disease.
A pure example of SaMD is the certified and successful app
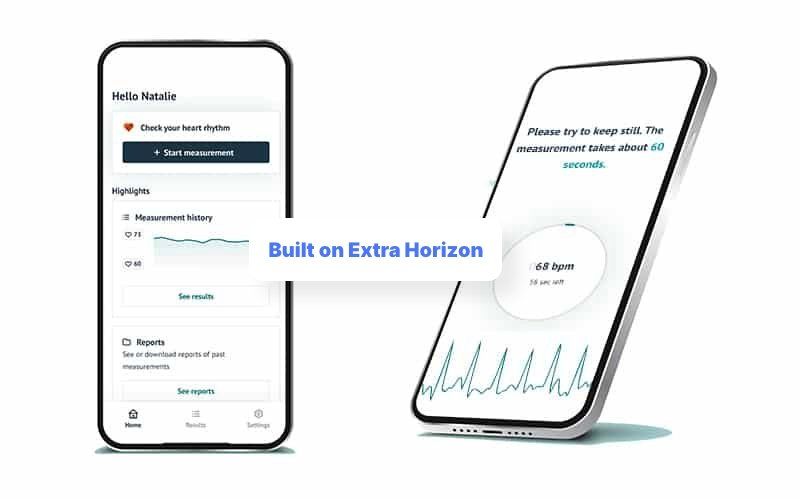
FibriCheck.
FibriCheck is a smartphone app that can check your heart rhythm, detecting any irregularities and therefore preventing strokes. FibriCheck is not part of a physical medical device, but is instead installed directly on the user’s smartphone. Almost any smartphone, that is! This is why FibriCheck is classified as SaMD.
SaMD offers many useful benefits. It can facilitate rapid data collection and analysis, which is far quicker and more efficient than what can be achieved by clinicians in the same timespan. It can also pick up on any sudden changes in a patient’s condition very quickly, even detecting subtle changes that may not have been noticed by the patient or their monitoring clinician. For this reason, SaMD can have considerable benefits to the patient, as detecting abnormal patterns early means that these can be investigated and addressed, thus opening the possibility of preventative care and preventing the patient from becoming more seriously ill.
Clinical evaluation for SaMD in EU markets
In essence, the purpose of performing clinical evaluation is to make sure that the SaMD conforms with the required regulatory requirements. In the process of clinical evaluation, the clinical data related to the SaMD is analysed and assessed to verify that the SaMD is safe to use and that the data is clinically useful. Performing clinical evaluation also enables any risks to be identified that have not already been identified and assessed as acceptable in the risk analysis, thus preventing any unexpected and potentially dangerous side effects.
Why do you need to perform clinical evaluation for SaMD (Software as a Medical Device?
In the EU, the path for clinical evaluation of SaMD is split into 5 stages. These are illustrated in the figure below:
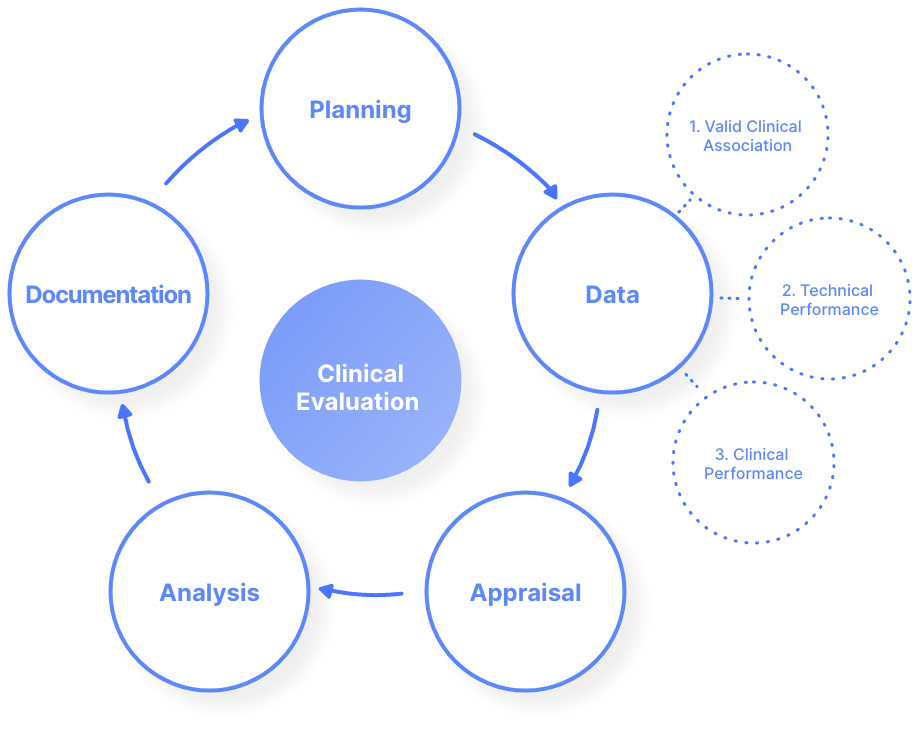
The requirements for the clinical evaluation of SaMD are outlined in Article 61 of the MDR. When it comes specifically to the clinical evaluation of SaMD, the path for this is outlined in the document ‘MDCG 2020-1 Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software’; a document endorsed by the Medical Device Coordination Group (MDCG) of the European Commission.
What is the MDR and the IVDR, and why do they matter when building SaMD?
The MDR (Medical Device Regulation) is a set of regulations that governs the manufacture and distribution of medical devices in the EU. It came into effect on 26 May 2021. The MDR aims to ensure a high quality and safety of medical devices in the EU.

The IVDR (In Vitro Medical Device Regulation) is a regulatory framework for in-vitro medical devices in the EU. It was entered into force on 26 May 2017, and will apply fully in all EU member states by 26 May 2022. The IVDR sets rules on how in-vitro medical devices are classified, and how they are regulated and kept under surveillance.
Both the MDR and the IVDR are especially important when building SaMD, as they are used as the basis of the requirements for building SaMD. Therefore, being compliant with the MDR and the IVDR is essential for any company developing SaMD.
The three main components in the EU clinical evaluation stage
The three main components in the EU SaMD clinical evaluation stage are as follows:
1. Valid Clinical Association of the SaMD
For this component, you must be able to prove a valid association between the output of your SaMD (e.g. measurements, conclusions) and the clinical condition that it is intended to target. You can prove a valid association by using things such as findings from clinical research and trials, secondary data analysis, etc.
2. Analytical / Technical Validation of the SaMD
Here, you must prove that your SaMD correctly processes input data to produce accurate and reliable output data. Not only must you prove that your SaMD processes data properly, but you must also show that your software has been built properly.
3. Clinical Validation
of the SaMD
For the clinical validation element, you must prove that any data produced by your SaMD is clinically meaningful and beneficial. For example, with the FibriCheck app, it must be proven that the data produced is clinically useful for detecting cardiac arrhythmias.
For more information on these three main components, you can read this document here.
There are many ways to obtain the necessary clinical data for the clinical evaluations of a SaMD. Here are several possible ways to collect the data:
- Published data on clinical experience with the device or equivalent
- Clinical investigation
- Clinical investigation with a similar device
- Combination of the above
How to improve the likelihood of passing the dreaded clinical evaluation phase for a SaMD
Your secret weapon for the Clinical Evaluation phase: The Lifecycle Tool
Extra Horizon offers a Lifecycle tool; a tool that makes it easier to manage software lifecycles. Lifecycle management refers to the specification, design, development, and testing of a software application, from the initial conception of the application, right up until the discontinuation of the product. The documentation generated by the Lifecycle Tool can be used as valuable evidence to prove that your SaMD conforms to the necessary regulatory requirements - something that is extremely important during the Clinical Evaluation Phase.
How Extra Horizon can help carry the burden of passing the Clinical Evaluation phase for your SaMD
Saving precious time and money
Using Extra Horizon’s medical Backend-as-a-Service (BaaS) offers numerous benefits during the Clinical Evaluation phase. As we provide a leveraged platform, this means that companies are able to simply configure their own backend platform instead of building it from scratch in-house. This will save you a lot of time and money, which means you will have more time and effort to focus on tackling the often-daunting SaMD Clinical Evaluation phase.
Regulatory certifications
Extra Horizon holds a number of regulatory certifications, including, but not limited to, the list below:
How does the SaMD clinical evaluation process differ in FDA-regulated markets?
In the FDA controlled regions, which includes the 50 United States, the District of Columbia, Puerto Rico, Guam, the Virgin Islands, American Samoa, and other U.S. territories and possessions, the clinical evaluation phase is quite similar to that of the EU markets, although there are some differences that are well worth mentioning. We're currently writing a piece that dives a bit deeper into this aspect, so keep your eyes open or stay in the loop via our newsletter.
To conclude
The Clinical Evaluation phase can be a very daunting process, but by paying close attention to the regulatory requirements and making sure that your SaMD has a good and thorough QMS, you will be able to pass the Clinical Evaluation phase with flying colours. By using Extra Horizon’s ready-made, already-compliant medical BaaS, you will relieve yourself of even more pressure during the process. To find out more, please do not hesitate to
get in touch.
RECENT POSTS

FREE EBOOKS


GOT QUESTIONS?